Journal of Chinese Pharmaceutical Sciences ›› 2022, Vol. 31 ›› Issue (12): 946-952.DOI: 10.5246/jcps.2022.12.080
• Original articles • Previous Articles Next Articles
Xintong Zhao, Yuhua Hu, Tong Qin, Tianlei Li, Wenxuan Zhang*( ), Song Wu*()
), Song Wu*()
Supporting:
1、Chemistry
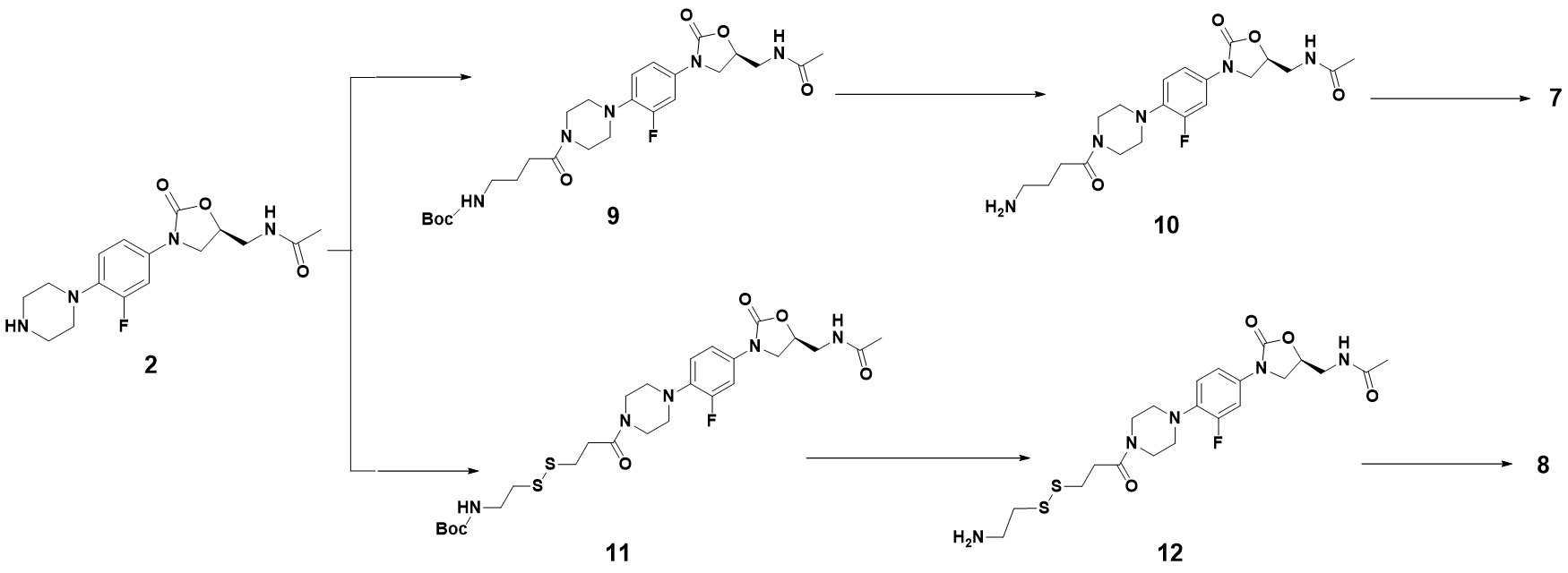
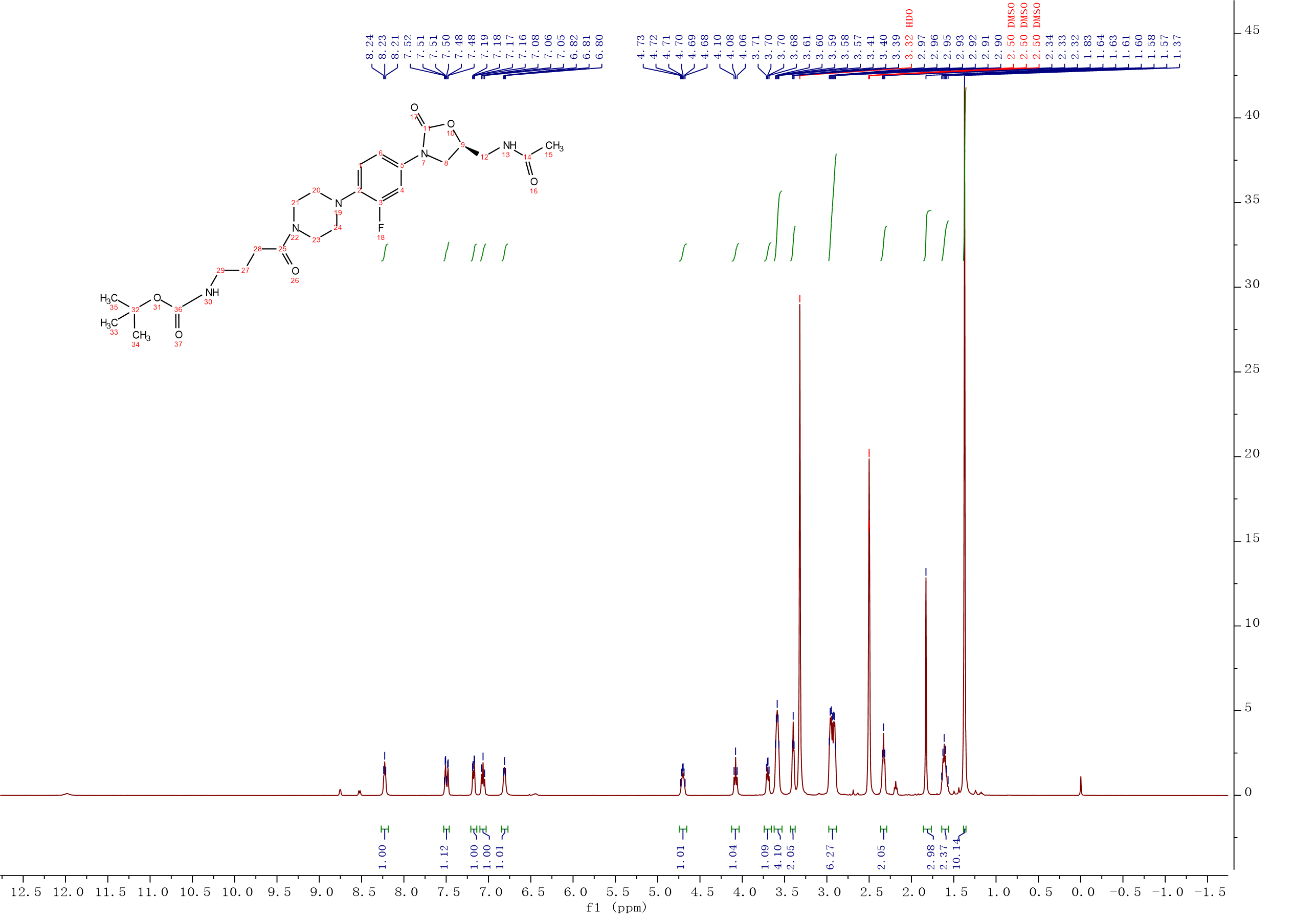
Intermediate 9:N-Boc-γ-aminobutyric acid (0.59g, 2.91 mmol), HATU (1.10g, 2.91 mmol), CH2Cl2 (5 mL) and Et3N (0.50 mL, 3.57 mmol) were added into the reaction flask, the solution was stirred at room temperature for 15 min. After oxazolidinone 2 (0.8 g, 2.38 mmol) was added, the solution was continuously stirred at room temperature for 4 h. The mixture was washed with 0.1M HCl solution, saturated NaHCO3 solution and saturated brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to afford 9 (1.10 g, 89%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.27 – 8.18 (m, 1H), 7.53 – 7.46 (m, 1H), 7.21 – 7.14 (m, 1H), 7.06 (t, J = 9.3 Hz, 1H), 6.84 – 6.77 (m, 1H), 4.75 – 4.66 (m, 1H), 4.08 (t, J = 8.9 Hz, 1H), 3.74 – 3.66 (m, 1H), 3.62 – 3.53 (m, 4H), 3.40 (t, J = 5.5 Hz, 2H), 2.98 – 2.89 (m, 6H), 2.33 (t, J = 7.4 Hz, 2H), 1.83 (s, 3H), 1.64 – 1.56 (m, 2H), 1.37 (s, 9H). 13C NMR (101 MHz, DMSO) δ 170.82 (CONH), 170.46 (CONH), 156.08 (C-36), 155.11 (C-3, d, JC-F = 242.0 Hz), 154.52 (C-11), 135.83 (Ar-C), 134.21 (Ar-C), 120.26 (Ar-C), 114.53 (Ar-C), 107.17 (Ar-C), 77.88 (C-32), 72.02 (C-9), 51.17 (CH2), 47.75 (2CH2), 45.40 (CH2), 41.87 (2CH2), 41.50 (CH2), 30.06 (C-28), 28.73 (C-33, C-34, C-35), 25.62 (C-27), 22.91 (C-15). ESI-MS calcd [M+Na]+ for C25H36FN5O6 544.2542, found 544.2550.
Intermediate 10:The intermediate 9 (1g, 1.92 mmol) and HCl/diethylether solution (2M, 6 mL, 9.58 mmol) were added into the reaction flask, the mixture was stirred at room temperature. After the completion of the reaction was monitored by the LC-MS, the solid was filtered and dried in vacuo overnight to afford compound 10 as an off-white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.32 – 8.23 (m, 1H), 7.94 (s, 3H), 7.54 – 7.46 (m, 1H), 7.24 – 7.15 (m, 1H), 7.14 – 7.03 (m, 1H), 4.76 – 4.65 (m, 1H), 4.08 (t, J = 9.0 Hz, 1H), 3.71 (s, 2H), 3.40 (d, J = 5.7 Hz, 3H), 3.21 (d, J = 10.2 Hz, 4H), 3.01 – 2.90 (m, 3H), 2.85 – 2.75 (m, 2H), 2.69 (s, 1H), 2.33 (t, J = 7.4 Hz, 1H), 1.83 (s, 3H), 1.81 – 1.76 (m, 1H). 13C NMR (101 MHz, DMSO) δ 170.53 (CONH), 170.30 (CONH), 154.53 (C-11), 155.10 (C-3, d, JC-F = 242.0 Hz), 135.70 (Ar-C), 134.28 (Ar-C), 120.32 (Ar-C), 114.58 (Ar-C), 107.21 (Ar-C), 72.00 (C-9), 51.11 (CH2), 47.78 (2CH2), 45.27 (CH2), 41.86 (2CH2), 41.55 (CH2), 30.93 (C-28), 29.68 (C-27), 22.91 (C-15). ESI-MS calcd [M+Na]+ for C20H28FN5O4 444.2018, found 444.2014.
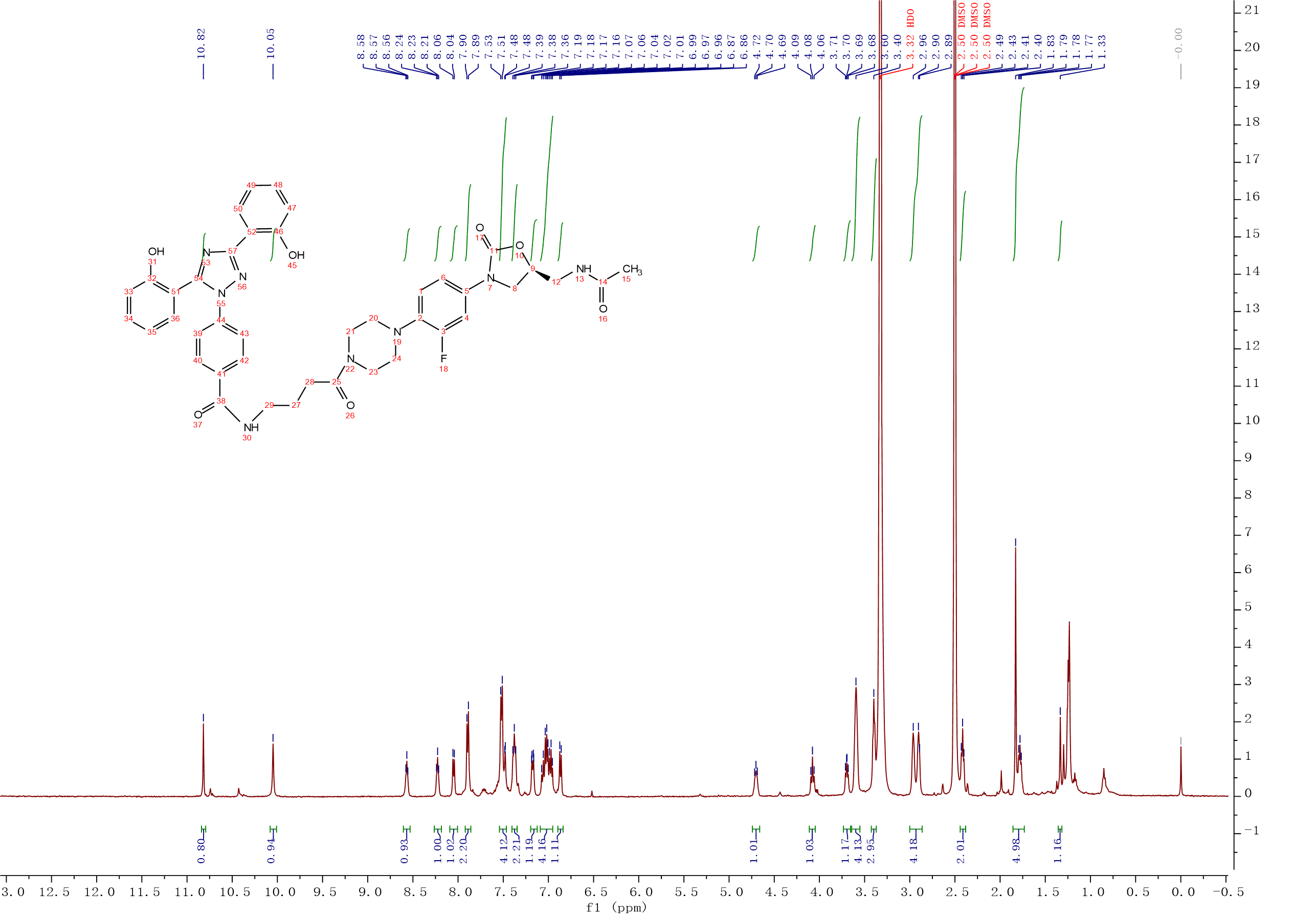
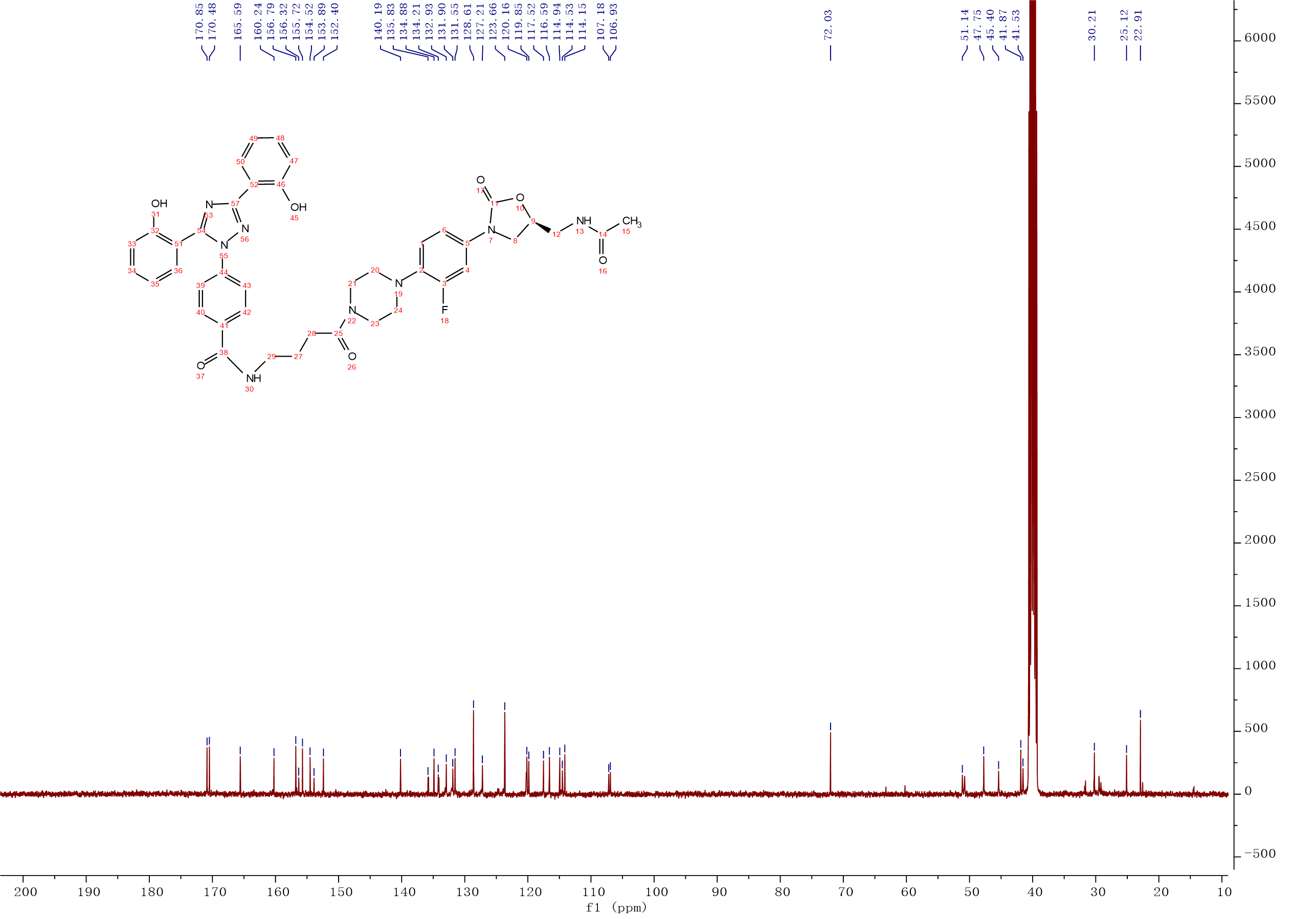
Compound 7:HATU (0.32g, 0.85 mmol) and Et3N (150 μL, 1.07 mmol) were added to a solution of deferasirox (0.32g, 0.85 mmol) in CH2Cl2 (5 mL), the solution was stirred at room temperature for 20min. After Intermediate 10 (0.3g, 0.71 mmol) was added, the solution was continuously stirred at room temperature for 3 h. The mixture was washed with 0.1M HCl solution, saturated NaHCO3 solution, saturated brine. The organic layer was dried over Na2SO4, filtered and concentrated, the crude product was purified by column chromatography with gradient elution (0-10% MeOH in CH2Cl2). Compound 7 (0.40g, 73%) was isolated as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.05 (s, 1H), 8.57 (t, J = 5.5 Hz, 1H), 8.23 (t, J = 5.9 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 8.1 Hz, 4H), 7.49 – 7.44 (m, 1H), 7.38 (t, J = 8.3 Hz, 1H), 7.20 – 7.12 (m, 1H), 7.04 (m, J = 14.8, 7.9 Hz, 3H), 6.97 (t, J = 7.9 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 4.71 (m, J = 7.4, 6.4 Hz, 1H), 4.08 (t, J = 8.9 Hz, 1H), 3.73 – 3.66 (m, 1H), 3.60 (s, 4H), 3.40 (s, 3H), 2.93 (d, J = 28.8 Hz, 4H), 2.41 (t, J = 7.4 Hz, 2H), 1.83 (s, 3H), 1.81 – 1.74 (m, 2H), 1.33 (s, 1H). 13C NMR (101 MHz, DMSO) δ 170.85 (CONH), 170.48 (CONH), 165.59 (C-38), 160.24 (Ar-C), 156.79 (Ar-C), 155.72 (Ar-C), 155.10 (C-3, d, JC-F = 243.0 Hz), 154.52 (C-11), 152.40 (Ar-C), 140.19 (Ar-C), 135.83 (Ar-C), 134.88 (Ar-C), 134.21 (Ar-C), 132.93 (Ar-C), 131.90 (Ar-C), 131.55 (Ar-C), 128.61 (2Ar-C), 127.21 (Ar-C), 123.66 (2Ar-C), 120.26 (Ar-C), 120.16 (Ar-C), 119.85 (Ar-C), 117.52 (Ar-C), 116.59 (Ar-C), 114.94 (Ar-C), 114.53 (Ar-C), 114.15 (Ar-C), 107.18 (Ar-C), 72.03 (C-9), 51.14 (CH2), 47.75 (2CH2), 45.40 (CH2), 41.87 (2CH2), 41.53 (CH2), 30.21 (C-28), 25.12 (C-27), 22.91 (C-15). ESI-MS calcd [M+Na]+ for C41H41FN8O7 799.2974, found 799.2973.
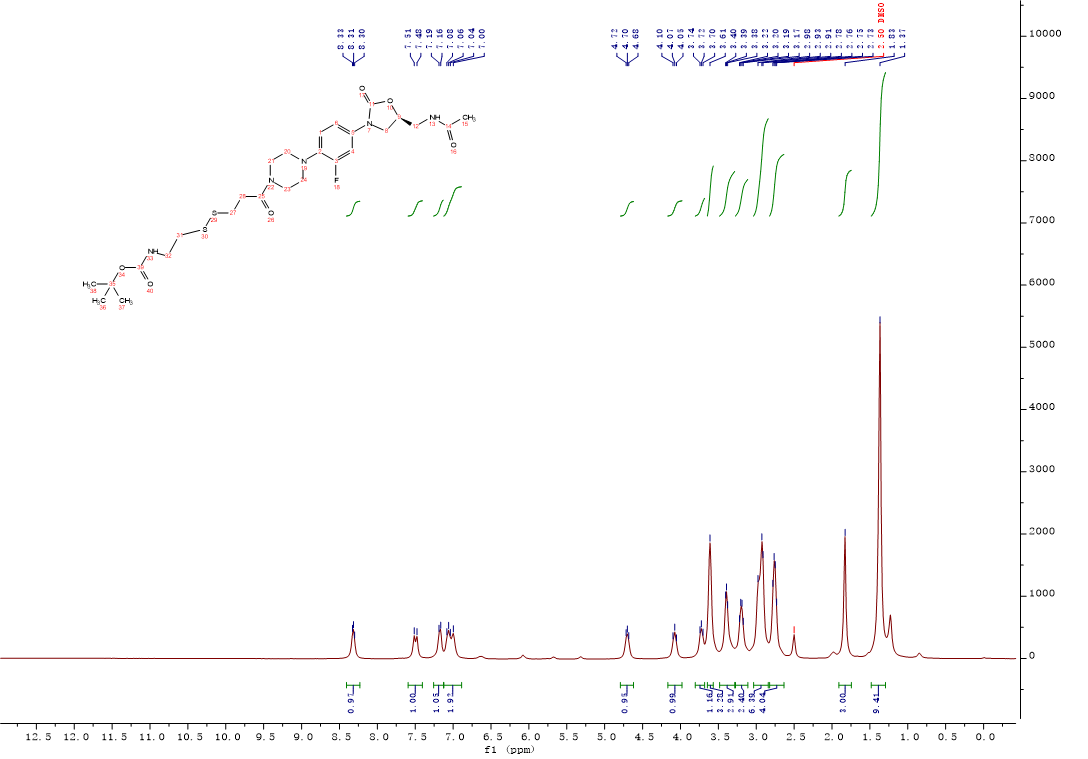
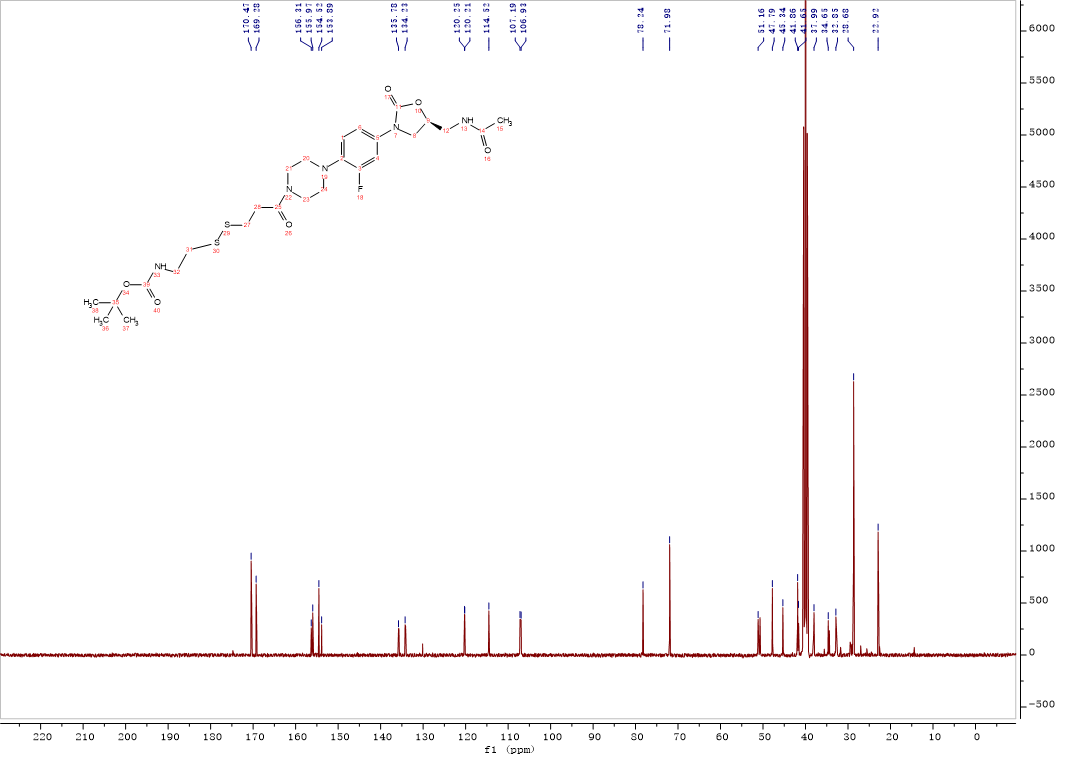
Intermediate 11:3-((2-((tert-Butoxycarbonyl)amino)ethyl)disulfanyl)propanoic acid (0.15g, 0.53 mmol), HATU (0.2g,0.53 mmol), dichloromethane (5 mL) and triethylamine (110 μL,0.80 mmol) were added into the reaction flask, the solution was stirred at room temperature for 15 min. After oxazolidinone 2 (0.23g, 0.68 mmol) was added, the solution was continuously stirred at room temperature overnight. The mixture was concentrated to remove CH2Cl2, and the residue was diluted with 0.1M HCl solution (10 mL) and extracted 2 times with n-butanol. The organic layer were combined and washed with saturated NaHCO3 solution and brine. The solvent was dried over Na2SO4, filtered and concentrated under reduced pressure to afford 11 (0.28g, 87%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.43 – 8.23 (m, 1H), 7.50 (d, J = 14.8 Hz, 1H), 7.26 – 7.13 (m, 1H), 7.12 – 6.89 (m, 2H), 4.79 – 4.62 (m, 1H), 4.17 – 3.98 (m, 1H), 3.72 (t, J = 7.9 Hz, 1H), 3.61 (s, 3H), 3.48 – 3.28 (m, 3H), 3.27 – 3.11 (m, 2H), 3.03 – 2.84 (m, 6H), 2.82 – 2.63 (m, 4H), 1.83 (s, 3H), 1.37 (s, 9H). 13C NMR (101 MHz, DMSO) δ 170.47 (CONH), 169.28 (CONH), 155.97 (C-39), 155.10 (C-3, d, JC-F = 242.0 Hz), 154.52 (C-11), 135.78 (Ar-C), 134.23 (Ar-C), 120.25 (Ar-C), 114.52 (Ar-C), 107.19 (Ar-C), 78.24 (C-35), 71.98 (C-9), 51.16 (CH2), 47.79 (2CH2), 45.34 (CH2), 41.86 (2CH2), 41.65 (CH2), 37.99 (CH2), 34.65 (CH2), 32.85 (CH2), 28.68 (C-36, C-37, C-38), 22.92 (C-15). ESI-MS calcd [M+Na]+ for C26H38FN5O6S2 622.2140, found 622.2157.
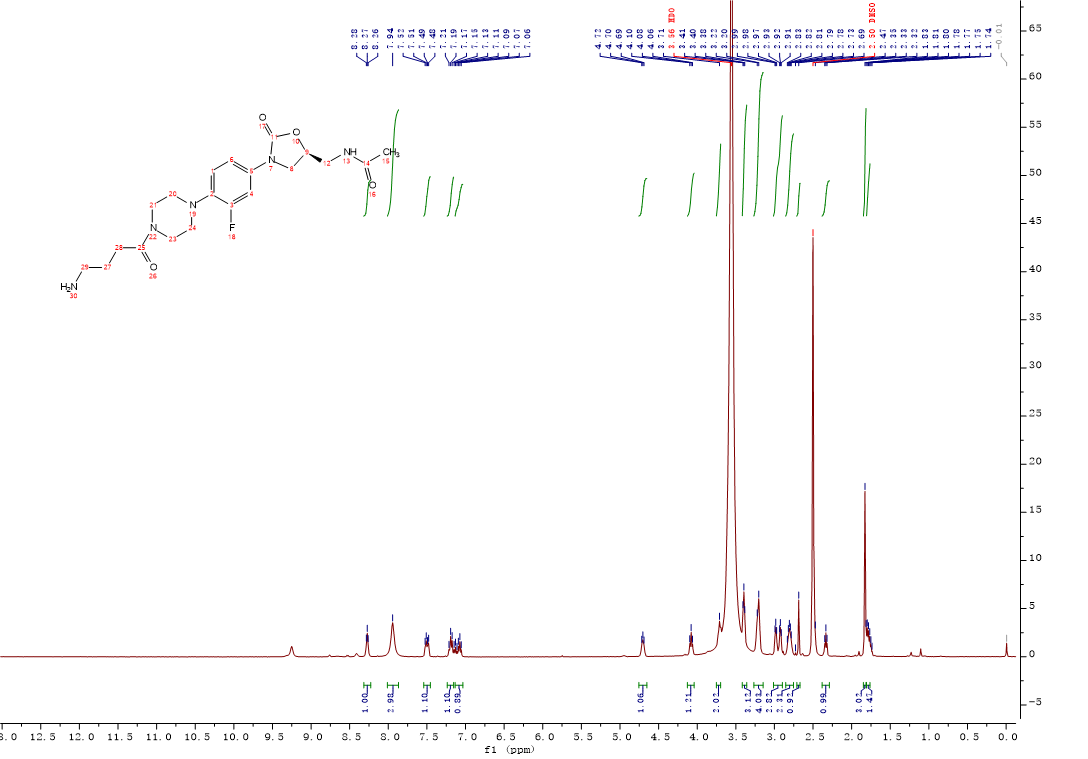
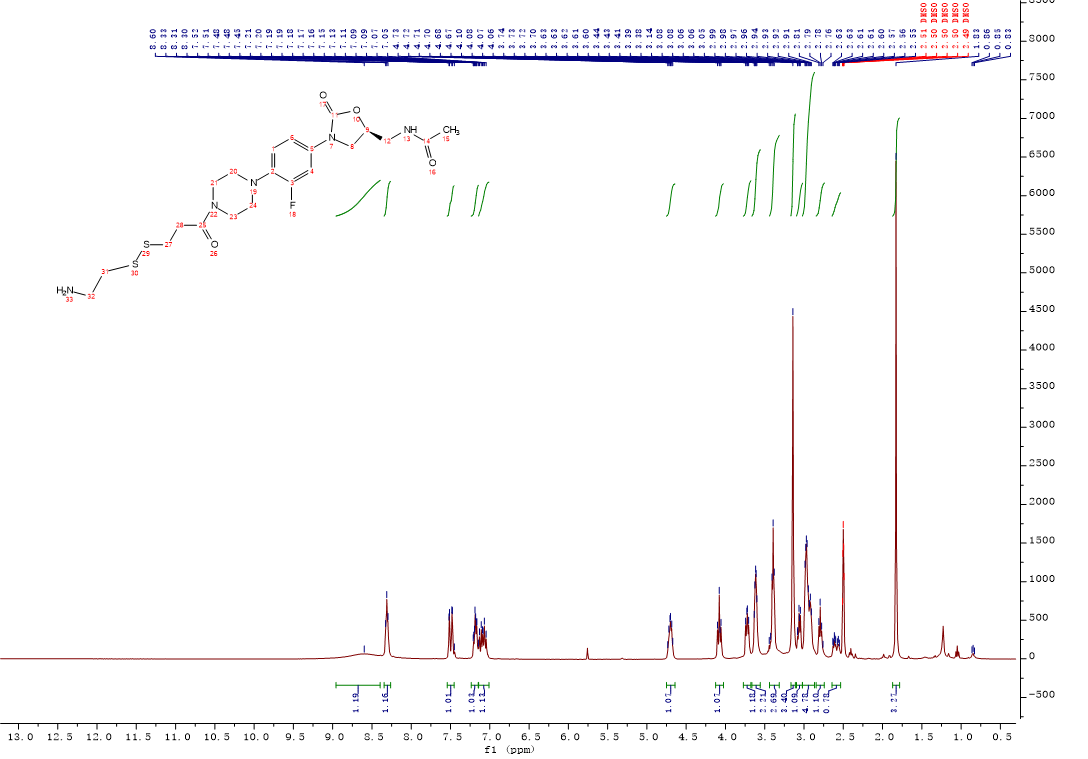
Intermediate 12:HCl/diethylether solution (2M, 1.2 mL, 2.3 mmol) was added to a solution of intermediate 11 (0.28g, 0.46 mmol) in CH2Cl2, the mixture was stirred at room temperature. After the completion of the reaction was monitored by the LC-MS, the mixture was concentrated under reduced pressure and dried in vacuo overnight to afford compound 12 (0.21g, 91%) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.31 (t, J = 5.9 Hz, 1H), 7.54 – 7.45 (m, 1H), 7.24 – 7.15 (m, 1H), 7.14 – 7.01 (m, 1H), 4.75 – 4.64 (m, 1H), 4.08 (t, J = 9.0 Hz, 1H), 3.77 – 3.68 (m, 1H), 3.66 – 3.56 (m, 2H), 3.39 (t, J = 5.6 Hz, 3H), 3.14 (s, 3H), 3.10 – 3.02 (m, 1H), 3.02 – 2.87 (m, 5H), 2.79 (t, J = 7.0 Hz, 1H), 2.64 – 2.53 (m, 1H), 1.83 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.47 (CONH), 169.29 (CONH), 155.10 (C-3, d, JC-F = 242.0 Hz), 154.52 (C-11), 135.79 (Ar-C), 134.49 (Ar-C), 120.27 (Ar-C), 114.54 (Ar-C), 107.21 (Ar-C), 72.00 (C-9), 50.70 (CH2), 47.78 (2CH2), 45.35 (CH2), 41.86 (2CH2), 41.67 (CH2), 38.40 (CH2), 34.59 (CH2), 32.82 (CH2), 22.92 (C-15). ESI-MS calcd [M+H]+ for C21H30FN5O4S2 500.1796, found 500.1791.
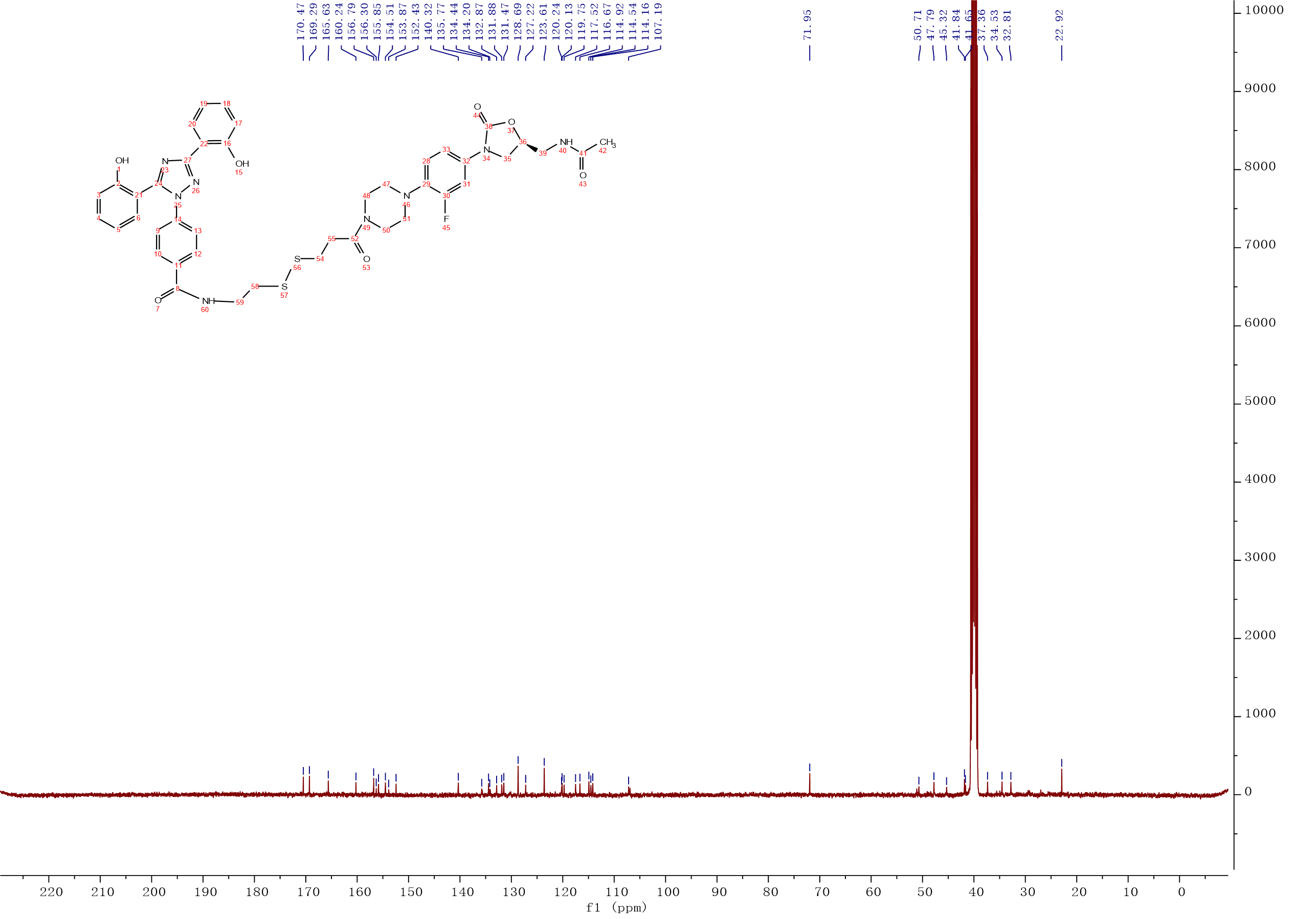
Compound 8:HATU (0.09 g, 0.24 mmol) and Et3N (42 μL,0.30 mmol) were added to a solution of deferasirox (0.09 g,0.24 mmol) in CH2Cl2 (3 mL), the solution was stirred at room temperature for 15 min. After intermediate 12 (0.1g,0.20 mmol) was added, the solution was continuously stirred at room temperature for 5 h. The mixture was washed with 0.1M HCl solution, saturated NaHCO3 solution, saturated brine. The organic layer was dried over Na2SO4, filtered and concentrated, the crude product was purified by column chromatography with gradient elution (0-10% MeOH in CH2Cl2). Compound 8 (0.14g, 84%) was isolated as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.10 (s, 1H), 8.81 (t, J = 5.6 Hz, 1H), 8.26 (t, J = 5.8 Hz, 1H), 8.04 (dd, J = 7.8, 1.6 Hz, 1H), 7.94 – 7.87 (m, 2H), 7.55 – 7.51 (m, 3H), 7.51 – 7.46 (m, 1H), 7.41 – 7.35 (m, 2H), 7.19 – 7.13 (m, 1H), 7.08 – 6.94 (m, 4H), 6.91 – 6.86 (m, 1H), 4.74 – 4.64 (m, 1H), 4.10 – 4.03 (m, 1H), 3.74 – 3.67 (m, 1H), 3.57 – 3.52 (m, 2H), 3.39 (t, J = 5.5 Hz, 3H), 3.00 – 2.88 (m, 9H), 2.77 (t, J = 7.0 Hz, 2H), 1.83 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.47 (CONH), 169.29 (CONH), 165.63 (C-8), 160.24 (Ar-C), 156.79 (Ar-C), 155.85 (Ar-C), 155.08 (C-30, d, JC-F = 243.0 Hz), 154.51 (C-38), 152.43 (Ar-C), 140.32 (Ar-C), 135.77 (Ar-C), 134.44 (Ar-C), 134.20 (Ar-C), 132.87 (Ar-C), 131.88 (Ar-C), 131.47 (Ar-C), 128.69 (2Ar-C), 127.22 (Ar-C), 123.61 (2Ar-C), 120.24 (Ar-C), 120.13 (Ar-C), 119.75 (Ar-C), 117.52 (Ar-C), 116.67 (Ar-C), 114.92 (Ar-C), 114.54 (Ar-C), 114.16 (Ar-C), 107.19 (Ar-C), 71.95 (C-36), 50.71 (CH2), 47.79 (2CH2), 45.32 (CH2), 41.84 (2CH2), 41.65 (CH2), 37.36 (CH2), 34.53 (CH2), 32.81 (CH2), 22.92 (C-15). ESI-MS calcd [M+Na]+ for C42H43FN8O7S2 877.2572, found 877.2571.
2、Minimum Inhibitory Concentration Testing
The minimum inhibitory concentrations (MICs) of the compounds were determined using an agar dilution method according to the methods of the National Committee for Clinical Laboratory Standards (NCCLS). All compounds were dissolved in a 50% aqueous solution of DMSO to prepare a stock solution with a concentration of 2560 μg/ mL. Serial 2-fold dilutions were prepared from the stock solutions using sterile water followed by a 10-fold dilution with CAMHB agar medium to provide concentration ranges of 256-0.125 μg/ mL. The tested organisms were grown in CAMHB broth medium at 35 ℃ for 8 h and were adjusted to a turbidity of a 0.5 McFarland standard. The compounds and bacteria solutions were dispensed into 96-well plate with total volume 200 μL, the mixture was then cultured for 18 h at 37 ℃. MICs of the compounds were determined using linezolid as positive controls.
Intermediate 9
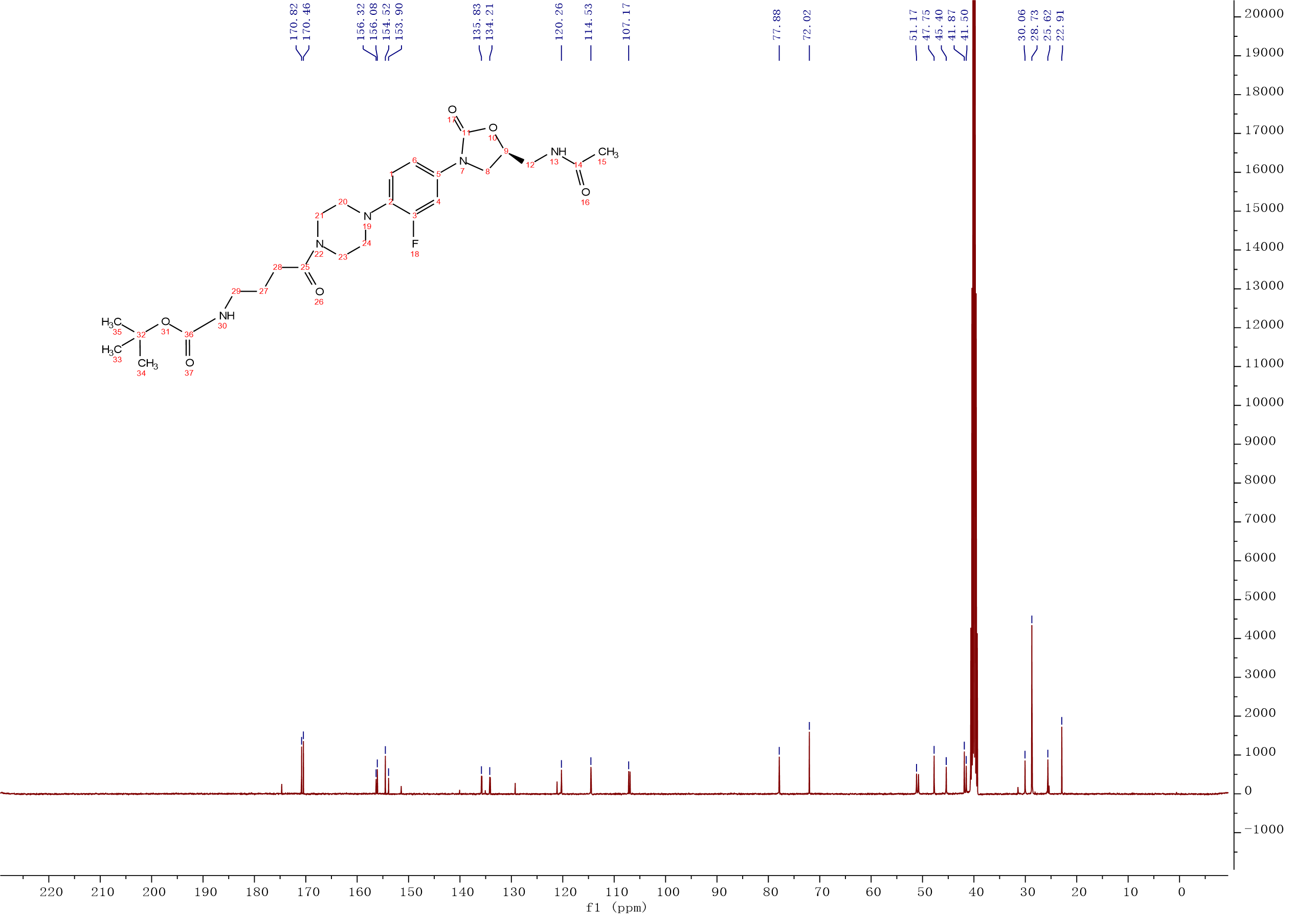
Intermediate 10
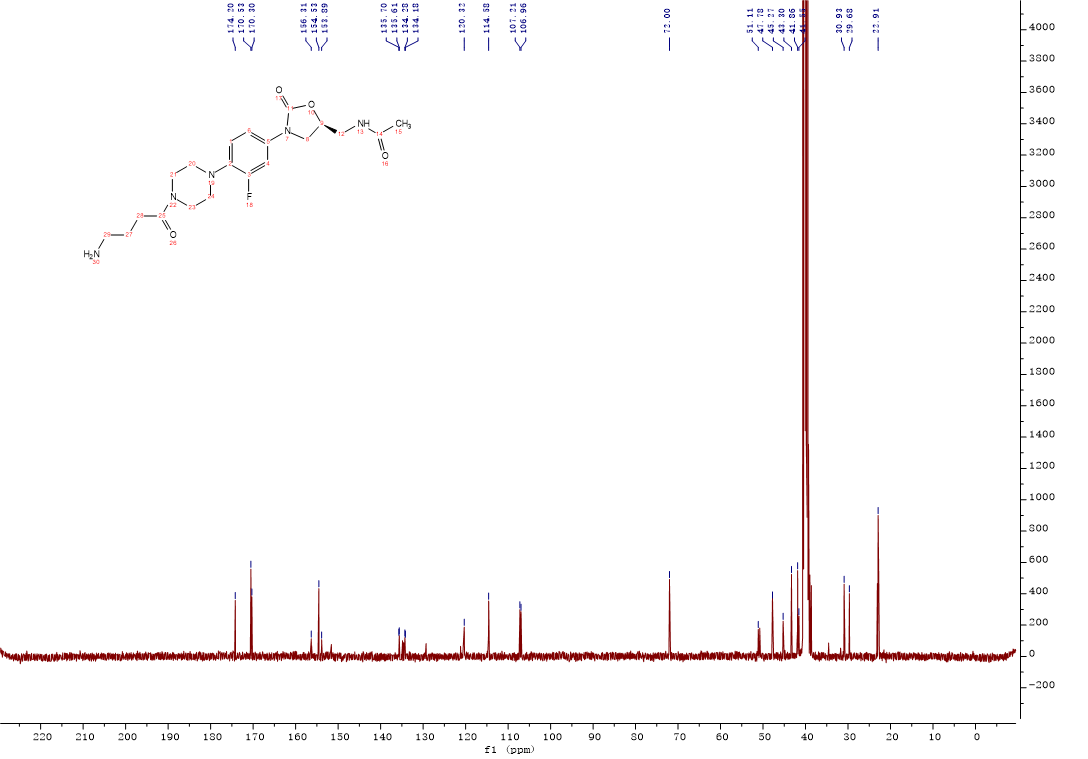
Compound 7
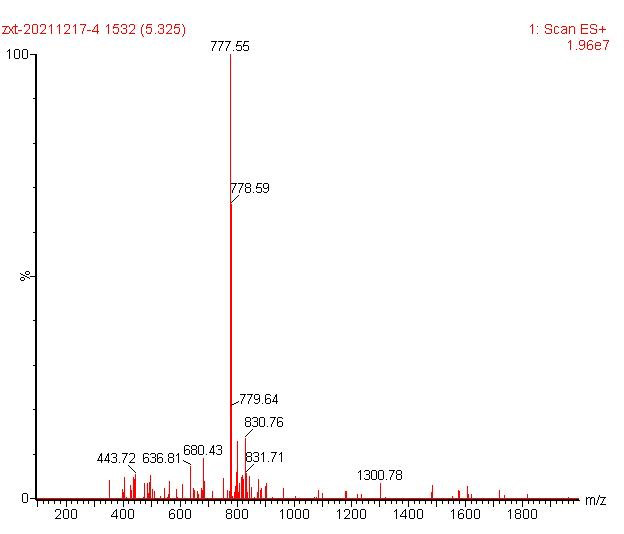
ESI-MS [M+H]+ 777.55
 #br#
#br#
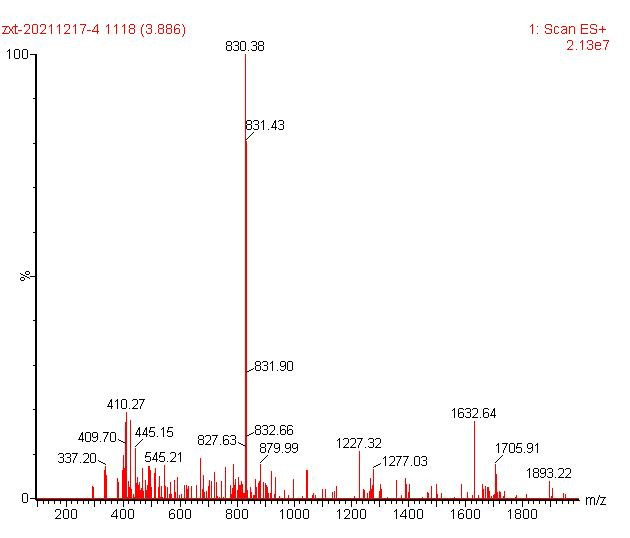
ESI-MS [M+Fe-2H]+ 830.38
Intermediate 11
Intermediate 12
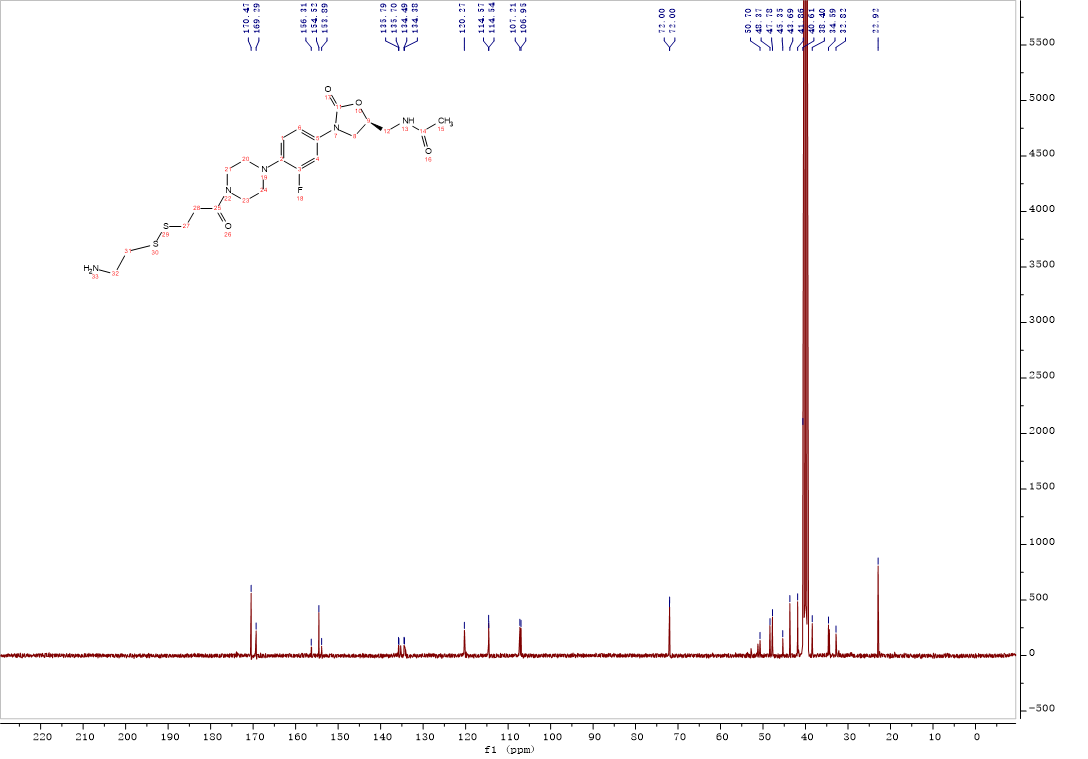
Compound 8

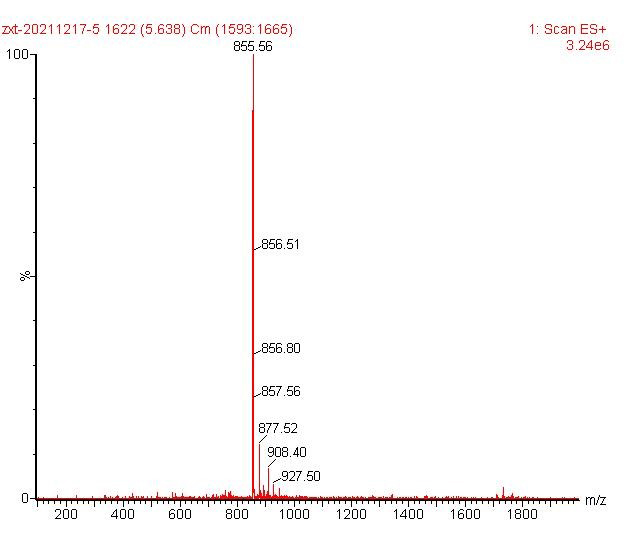
ESI-MS [M+H]+ 855.56
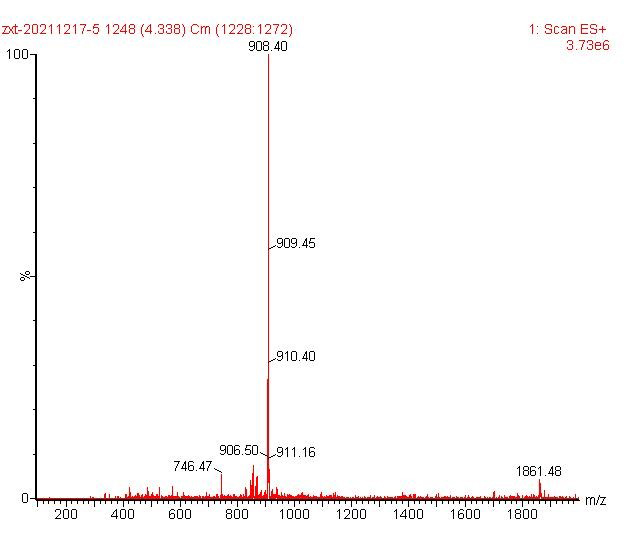
ESI-MS [M+Fe-2H]+ 908.40
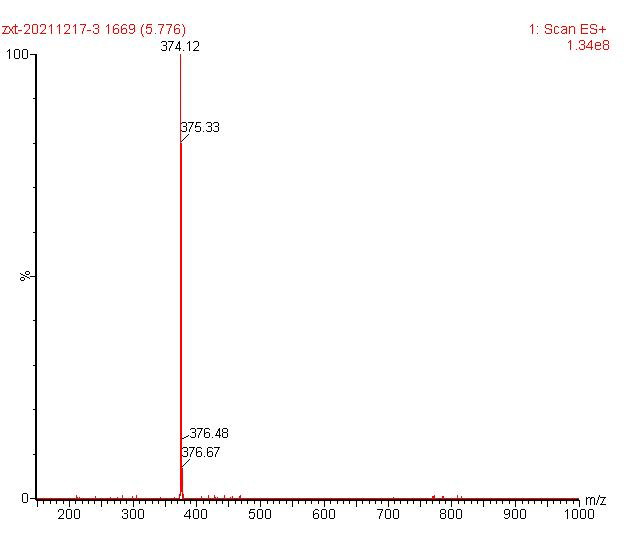
Deferasirox ESI-MS [M+H]+ 374.12
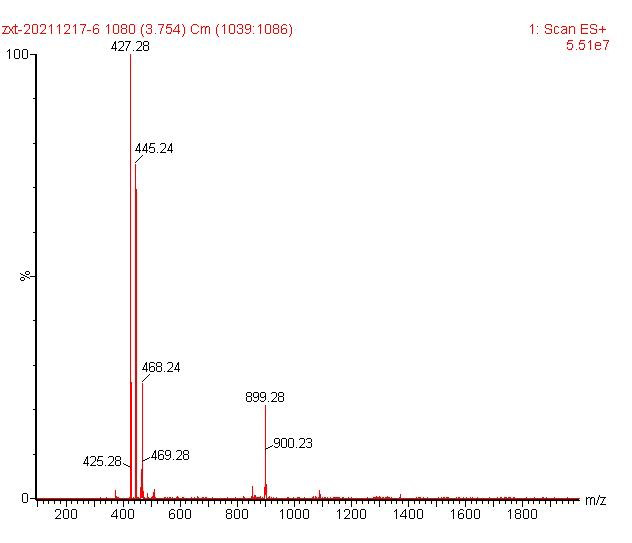
ESI-MS [M+Fe-2H]+ 427.28

 >
> >
>12-946/img_1.jpg)
12-946/img_2.jpg)
12-946/img_3.jpg)
12-946/img_4.jpg)
12-946/img_5.jpg)